
Feb 23
/
Antonia von Doctorflix
Hämoglobinopathien – Was du als Hausarzt wissen musst
Hämoglobinopathien wie Sichelzellkrankheit und Thalassämien sind weltweit verbreitet und betreffen zunehmend auch europäische Patienten. Erfahre als Hausarzt, wie du diese Erkrankungen frühzeitig erkennst, diagnostizierst und behandelst.
1. Einführung
Hämoglobinopathien gehören mit etwa 7 % Anlageträgern 1 zu den weltweit häufigsten genetischen Erkrankungen. Diese Krankheiten, wie die Sichelzellkrankheit und Thalassämien, entstehen durch genetische Veränderungen des Hämoglobins und betreffen Millionen von Menschen. Sie sind nicht nur in endemischen Regionen wie Afrika, dem Mittelmeerraum oder Asien von Bedeutung, sondern auch in Europa und Nordamerika durch Migration immer häufiger anzutreffen.2
Die Folgen dieser Erkrankungen sind weitreichend: Neben den gesundheitlichen Herausforderungen leiden viele Betroffene unter psychischen und sozialen Folgen. Familien sind oft durch hohe emotionale und finanzielle Belastungen beeinträchtig. Die weltweit steigende Prävalenz durch Migration und bessere Diagnostik bedeutet, dass diese Erkrankungen zunehmend auch in unserem Gesundheitssystem, das primär nicht davon betroffen war, eine Rolle spielen.
Ein frühes Erkennen von Hämoglobinopathien ist essenziell, um geeignete Therapien einzuleiten und Komplikationen vorzubeugen. Beispielsweise kann ein Neugeborenenscreening bei Kindern mit Migrationshintergrund eine Sichelzellkrankheit frühzeitig diagnostizieren und so schwerwiegende Komplikationen wie Schlaganfälle oder Infektionen verhindern. Ein weiteres Szenario ist die rechtzeitige Diagnosestellung bei Jugendlichen mit unerklärter Anämie, was eine gezielte Behandlung und Beratung möglich macht. Als Haus- oder Kinderarzt nimmst du dabei eine zentrale Rolle ein, von der initialen Diagnosestellung über die Einleitung von Spezialbehandlungen bis hin zur langfristigen Betreuung. Darüber hinaus kannst du durch gezielte Aufklärung der Patienten und ihrer Familien dazu beitragen, den Umgang mit der Krankheit zu verbessern.
2. Definition und Pathophysiologie
Hämoglobinopathien sind genetische Erkrankungen, die das Hämoglobin in roten Blutkörperchen betreffen. Die Hauptgruppen sind:
- Sichelzellkrankheit: Bei dieser Erkrankung sorgt eine genetische Mutation im HbB-Gen dafür, dass sich das Hämoglobin (HbS) verändert. Wenn zu wenig Sauerstoff vorhanden ist, verklumpt das HbS, und die roten Blutkörperchen verformen sich zu einer Sichelform. Das kann die Durchblutung behindern und Komplikationen auslösen.
- Thalassämien: Bei Thalassämien werden Alpha- oder Beta-Globinketten entweder gar nicht oder nur vermindert produziert. Dadurch gerät das Hämoglobin aus dem Gleichgewicht, was die Stabilität und die Funktion der roten Blutkörperchen beeinträchtigt.
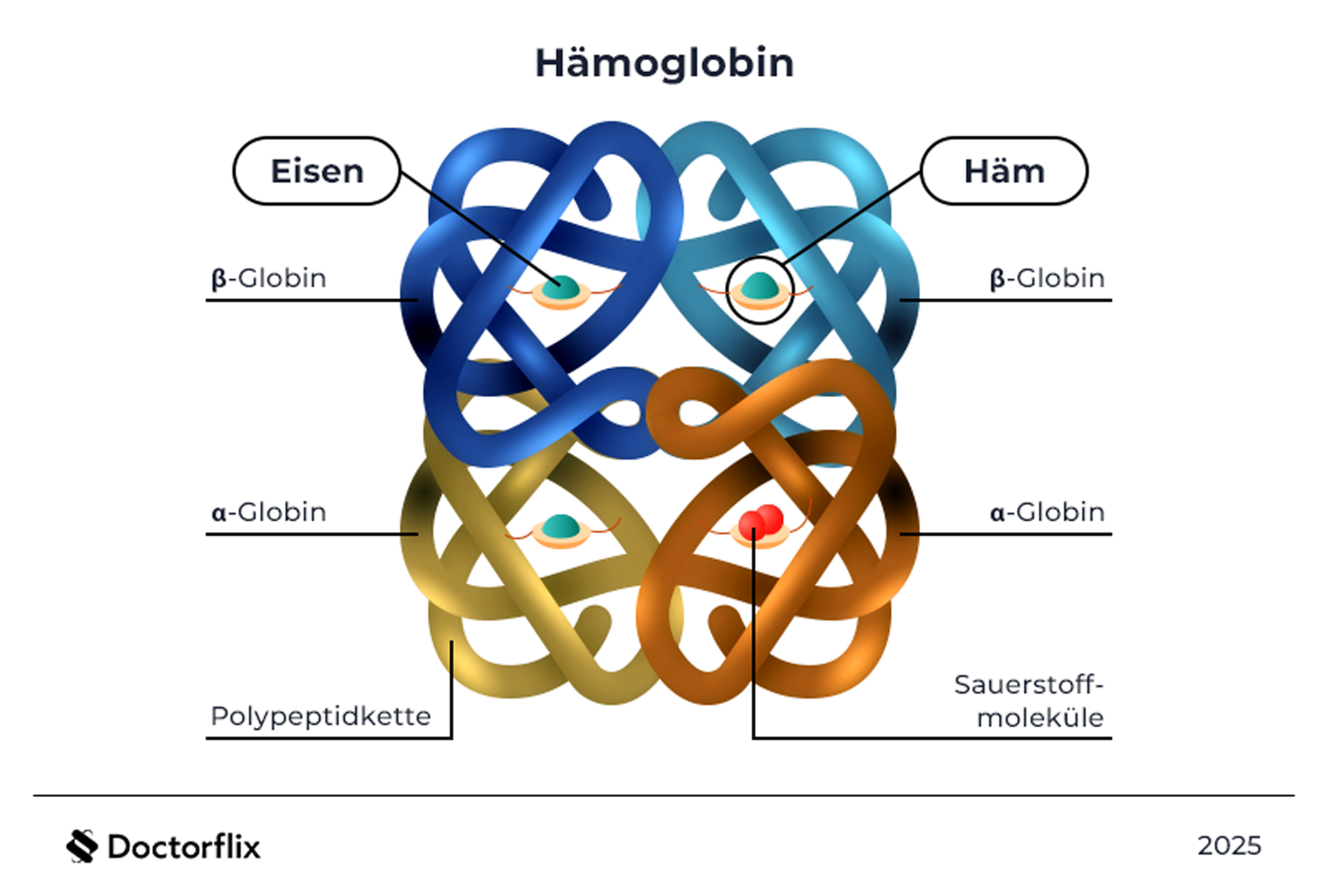
Genetische Ursachen und Pathomechanismen
Hämoglobinopathien werden in der Regel autosomal-rezessiv vererbt. Bei heterozygoten Trägern (z. B. Sichelzelltrait) treten meist keine oder nur milde Symptome auf, wie gelegentliche Belastungsintoleranz oder leichte Anämie unter extremen Bedingungen wie hoher Höhe oder intensiver sportlicher Betätigung. Risikofaktoren wie Dehydratation oder Sauerstoffmangel können jedoch in seltenen Fällen Symptome auslösen.3 Homozygote oder compound-heterozygote Formen hingegen können schwere Krankheitsbilder hervorrufen.
Ein zentraler Pathomechanismus bei der Sichelzellkrankheit ist die Verklumpung von HbS unter Sauerstoffmangel, wodurch die Zellmembran geschädigt wird und die Flexibilität der roten Blutkörperchen verloren geht. Dies führt zu sogenannten vaso-okklusiven Krisen, bei denen die verformten Zellen kleine Blutgefäße blockieren. Dadurch wird die Sauerstoffversorgung des umliegenden Gewebes gestört, was starke Schmerzen, Schwellungen und in schweren Fällen auch Gewebeschäden verursachen kann. Besonders betroffen sind Hände, Füße, Brustkorb und Bauch. Ohne schnelle Behandlung können solche Krisen lebensbedrohlich werden.
Thalassämien hingegen führen zu einer ineffizienten Erythropoese (Bildung der roten Blutkörperchen) und einer erhöhten Apoptose (programmierter Zelltod) von Vorläuferzellen im Knochenmark. Dies resultiert in einer verminderten Anzahl reifer roter Blutkörperchen und einer chronischen Anämie. Um den Sauerstoffmangel zu kompensieren, wird die Hämatopoese (Blutbildung) verstärkt, was zu einer Erweiterung des Knochenmarks und einer extramedullären (außerhalb des Knochenmarks) Blutbildung führen kann. Dies wiederum verursacht typische Knochenveränderungen wie eine verdickte Schädelkalotte oder Deformitäten im Gesicht („Chipmunk Face“).
Unterschiede zwischen Sichelzellkrankheiten und Thalassämien
Die beiden Erkrankungen unterscheiden sich deutlich:
- Sichelzellkrankheiten machen sich vor allem durch vaso-okklusive Krisen, starke Schmerzen und Organschäden bemerkbar.4
- Thalassämien führen zu schwerer Anämie, Skelettveränderungen und Komplikationen durch Eisenüberladung.5
Kombinierte Formen
Kombinierte Formen treten auf, wenn ein Patient Mutationen für mehrere Hämoglobinopathien trägt, wie etwa HbS/ß-Thalassämie. Solche Konstellationen verschlimmern oft die klinische Symptomatik. Patienten mit HbS/ß-Thalassämie zeigen beispielsweise sowohl vaso-okklusive Episoden als auch Symptome einer Thalassämie-bedingten Anämie.6
4. Symptome
Symptome bei Kindern
Kinder mit Hämoglobinopathien entwickeln oft im ersten Lebensjahr Symptome.
Bei Thalassämien manifestieren sich die Symptome in Form von schwerer Anämie, die durch Bluttransfusionen behandelt werden müssen. Ein charakteristisches Merkmal ist das veränderte Gesichtsprofil („chipmunk face“) durch Knochenmarkhyperplasie.
Bei der Sichelzellkrankheit treten vaso-okklusive Krisen auf, die zu Schmerzen, Schwellungen (besonders in den Händen und Füßen) und erhöhter Infektanfälligkeit führen. Weitere Symptome umfassen:
- Verzögertes Wachstum und spätes Einsetzen der Pubertät
- Milzinfarkte und funktionelle Asplenie
- Erhöhte Mortalität durch bakterielle Infektionen
Bei Thalassämien manifestieren sich die Symptome in Form von schwerer Anämie, die durch Bluttransfusionen behandelt werden müssen. Ein charakteristisches Merkmal ist das veränderte Gesichtsprofil („chipmunk face“) durch Knochenmarkhyperplasie.
Symptome bei Erwachsenen
Erwachsene erleben oft chronische Beschwerden wie Schmerzen, Fatigue und Organschäden. Typische Organschäden umfassen Herzprobleme, wie Kardiomyopathie durch Eisenüberladung, und Leberfunktionsstörungen, die durch chronische Transfusionen und Eisenakkumulation ausgelöst werden. Bei Thalassämien kann eine langjährige Transfusionstherapie zu schwerer Eisenüberladung und daraus resultierenden Herz- und Leberkomplikationen führen. Zusätzlich treten häufig psychosoziale Probleme wie Depressionen auf.
5. Komplikationen und ihre Behandlung
Komplikationen im Kindesalter
Kinder mit Hämoglobinopathien sind anfällig für:
- Schlaganfälle: Schlaganfälle sind eine ernste Gefahr für Kinder mit Sichelzellänämie – etwa 10 % von ihnen erleiden vor ihrem 20. Geburtstag einen Schlaganfall. Damit das nicht passiert, musst du als Arzt schnell handeln: Frühzeitig die Diagnose stellen und sofort eine Austauschtransfusion einleiten, um bleibende Schäden zu vermeiden.
- Infektionen: Kinder mit funktioneller Asplenie bzw. Splenomegalie sind besonders anfällig für Infektionen, vor allem durch Erreger wie Meningokokken, Pneumokokken und Haemophilus influenzae. Um schwere Verläufe zu vermeiden, solltest du auf vorbeugende Maßnahmen wie eine Penicillintherapie und wichtige Impfungen achten. Wenn dir ein Kind vorgestellt wird und du den Verdacht auf eine Infektion hast, dann zögere nicht – sorge sofort für eine Behandlung mit intravenösen Antibiotika.
- Chronische Anämie: Das klingt harmlos, kann aber gravierende Folgen haben. Kinder wachsen langsamer, können sich schlechter konzentrieren, und der Körper versucht verzweifelt, den Sauerstoffmangel auszugleichen. Dabei vergrößern sich oft Leber und Milz, und es kommt zu Verdickungen der Knochen, besonders im Kopf und Gesicht. Achte darauf, solche Anzeichen frühzeitig zu erkennen!
- Schmerzkrisen: Diese Krisen sind die häufigsten Notfälle bei der Sichelzellkrankheit und entstehen durch verstopfte Blutgefäße. Die Schmerzen sind stark, halten mindestens zwei Stunden an und treten oft in Armen, Beinen, Rücken oder Bauch auf. Wenn du mit so einem Fall konfrontiert bist, denk daran: schnelle Hilfe ist gefragt! Eine gute Schmerztherapie mit Morphin und ausreichend Flüssigkeit, am besten intravenös, kann die Durchblutung wieder verbessern und die Schmerzen lindern.
- ZNS-Infarkte: Dabei kommt es zu Durchblutungsstörungen im Gehirn, die sich durch neurologische Ausfälle wie Lähmungen oder Sprachprobleme bemerkbar machen können. Wenn du den Verdacht auf einen solchen Infarkt hast, ist schnelles Handeln entscheidend: Lass sofort eine Bildgebung machen, um die Diagnose zu sichern, und leite eine Austauschtransfusion ein, um größere Schäden zu verhindern.
- Priapismus: Wenn diese schmerzhafte Schwellung der Schwellkörper plötzlich auftritt, heißt es schnell handeln! Sorge für eine gute Schmerzlinderung und stelle sicher, dass der Betroffene ausreichend Flüssigkeit bekommt. Wenn das allein nicht reicht, zögere nicht, einen Urologen hinzuzuziehen, um größere Komplikationen zu vermeiden.
- Girdle-Syndrom: Bei diesem Syndrom, das durch Gefäßinfarkte im Darmbereich entsteht, kommt es zu Symptomen wie einem geblähten Bauch und fehlenden Darmgeräuschen. Wenn du solche Anzeichen siehst, solltest du sofort handeln: Eine Transfusion ist notwendig, um die Patienten zu stabilisieren und weitere Komplikationen zu vermeiden.
- Akute Milzsequestration: Hierbei staut sich Blut in der Milz, was zu einem hypovolämischen Schock führen kann. Du solltest sofort handeln: Vorsichtige Erythrozytenkonzentrats-Transfusionen sind notwendig, um eine Hyperviskosität zu vermeiden. Achte auch darauf, die Milzgröße regelmäßig zu kontrollieren, damit ein Fortschreiten rechtzeitig erkannt wird.
EXKURS AKUTES THORAXSYNDROM
Wenn du den Verdacht auf ATS hast, ist schnelles Handeln gefragt: Sorge dafür, dass der Betroffene Sauerstoff bekommt, und starte eine intravenöse Flüssigkeitszufuhr, um die Durchblutung zu unterstützen. Gleichzeitig ist es absolut notwendig, die Person sofort ins Krankenhaus zu bringen, am besten in eine Klinik mit intensivmedizinischer Versorgung. Dort können weiterführende Behandlungen wie eine Austauschtransfusion durchgeführt werden.
Um solche Notfälle möglichst zu vermeiden, ist es wichtig, deine Patienten und deren Familien über die Warnzeichen zu informieren. Präventive Maßnahmen wie Impfungen und regelmäßige Kontrolluntersuchungen helfen, Risiken zu minimieren und rechtzeitig einzugreifen.
Wichtige Hinweise:
Falls du weiterführende Informationen zu den möglichen Komplikationen bei Kindern haben möchtest, kannst du die kostenlose Fortbildung auf Doctorflix zum Thema "Pädiatrische Notfälle bei der Versorgung von Patienten mit Sichelzellkrankheit" gucken und dabei CME Punkte sammeln.
Das Akute Thoraxsyndrom (ATS) ist eine ernste und potenziell lebensgefährliche Komplikation, die bei Menschen allen Alters mit Sichelzellkrankheit auftreten kann, bei Kindern jedoch am häufigsten auftritt und eine Sterblichkeitsrate bis zu 10% haben kann.7,8 Es zeigt sich durch plötzliche Atemnot, Husten, Brustschmerzen und Fieber. Auch wenn der Sauerstoffgehalt im Blut absinkt oder du im Röntgenbild Veränderungen in der Lunge siehst, solltest du sofort hellhörig werden.
Wenn du den Verdacht auf ATS hast, ist schnelles Handeln gefragt: Sorge dafür, dass der Betroffene Sauerstoff bekommt, und starte eine intravenöse Flüssigkeitszufuhr, um die Durchblutung zu unterstützen. Gleichzeitig ist es absolut notwendig, die Person sofort ins Krankenhaus zu bringen, am besten in eine Klinik mit intensivmedizinischer Versorgung. Dort können weiterführende Behandlungen wie eine Austauschtransfusion durchgeführt werden.
Um solche Notfälle möglichst zu vermeiden, ist es wichtig, deine Patienten und deren Familien über die Warnzeichen zu informieren. Präventive Maßnahmen wie Impfungen und regelmäßige Kontrolluntersuchungen helfen, Risiken zu minimieren und rechtzeitig einzugreifen.
Wichtige Hinweise:
- Frühzeitige Erkennung und Behandlung: Dies ist entscheidend, um lebensbedrohliche Folgen zu verhindern.
- Infektionsmanagement: Jede Infektion sollte ernst genommen und bei Bedarf mit intravenösen Antibiotika behandelt werden.
- Prophylaxe: Maßnahmen wie eine Penicillinprophylaxe und Impfungen sind essenziell, um schwere Infektionen zu vermeiden.
Falls du weiterführende Informationen zu den möglichen Komplikationen bei Kindern haben möchtest, kannst du die kostenlose Fortbildung auf Doctorflix zum Thema "Pädiatrische Notfälle bei der Versorgung von Patienten mit Sichelzellkrankheit" gucken und dabei CME Punkte sammeln.
Komplikationen im Erwachsenenalter
Erwachsene sind oft von chronischem Organversagen betroffen. Zu den häufigen Komplikationen gehören:
- Niereninsuffizienz: Bei Menschen mit Sichelzellkrankheit können die Nieren durch wiederholte, vaso-okklusive Schäden und Hyperfiltration beeinträchtigt werden. Achte auf Anzeichen wie anhaltende Proteinurie oder eine verminderte Nierenfunktion und leite gegebenenfalls frühzeitig nephrologische Betreuung ein.
- Kardiomyopathie: Diese schwere Herzschädigung entsteht durch Eisenablagerungen, die bei Patienten mit chronischen Bluttransfusionen auftreten können. Du solltest regelmäßig den Eisenstatus überwachen und bei Anzeichen von Überladung sofort eine Chelattherapie einleiten, um das Herz zu entlasten und weitere Komplikationen zu vermeiden.
- Psychische Belastungen: Patienten mit chronischen Schmerzen und sozialer Isolation sind besonders anfällig für Depressionen. Diese Belastungen können die Lebensqualität stark beeinträchtigen, daher ist es wichtig, frühzeitig psychologische Unterstützung anzubieten oder an spezialisierte Fachkräfte zu überweisen.
6. Therapieansätze
Klassische Therapien
Achtung: Dieser Artikel ist für medizinische Fachkräfte, Ärztinnen und Ärzte und stellt keine Behandlungsempfehlung dar. Therapieentscheidungen werden nur von Ärzten aufgrund der individuellen Patientensituation getroffen. Im Zweifel konsultiere einen Facharzt.
Ausführlich werden die Therapiemethoden in drei unserer CME-Fortbildungen behandelt. Diese sind für dich am Ende dieses Artikels verlinkt.
- Transfusionen: Lebensrettend bei schwerer Anämie, jedoch mit dem Risiko der Eisenüberladung. Die regelmäßige Überwachung des Eisenstatus und eine begleitende Chelattherapie sind essenziell, um langfristige Organschäden zu vermeiden.
- Hydroxyharnstoff: Dieses Medikament ist insbesondere bei Sichelzellpatienten wirksam. Es fördert die Produktion von fetalem Hämoglobin (HbF), welches die Bildung von Sichelzellen hemmt. Dadurch werden vaso-okklusive Krisen reduziert und die Lebensqualität verbessert.
- Chelattherapie: Eine wirksame Maßnahme zur Behandlung von Eisenüberladung, die durch häufige Transfusionen entsteht. Moderne Chelatbildner, wie Deferasirox, ermöglichen eine orale Einnahme und verbessern die Compliance.
Ausführlich werden die Therapiemethoden in drei unserer CME-Fortbildungen behandelt. Diese sind für dich am Ende dieses Artikels verlinkt.
Fortschritte in der Gentherapie
Die “Genschere” CRISPR/Cas9 ermöglicht die Korrektur genetischer Defekte. Erste Studien zeigen, dass einige Patienten nach einer Behandlung ohne Transfusion auskamen. Allerdings gibt es auch Herausforderungen und Risiken bei dieser Therapie, wie die mögliche Off-Target-Editierung, die unerwünschte genetische Veränderungen verursachen kann. Zudem stehen die Langzeitfolgen der Behandlung noch nicht abschließend fest, und die Kosten sowie die komplexe Durchführung schränken derzeit die breite Verfügbarkeit ein.9,10
Ein großes Hindernis für die Anwendung der Gentherapie in endemischen Ländern ist die eingeschränkte Infrastruktur. Die Durchführung der Therapie erfordert spezialisierte Labore, qualifiziertes Personal und eine kontinuierliche Nachsorge, die in vielen Ländern des globalen Südens nicht allerorts verfügbar sind. Diese Diskrepanz zwischen entwickelten und weniger entwickelten Ländern stellt eine große globale Herausforderung dar.
Ein großes Hindernis für die Anwendung der Gentherapie in endemischen Ländern ist die eingeschränkte Infrastruktur. Die Durchführung der Therapie erfordert spezialisierte Labore, qualifiziertes Personal und eine kontinuierliche Nachsorge, die in vielen Ländern des globalen Südens nicht allerorts verfügbar sind. Diese Diskrepanz zwischen entwickelten und weniger entwickelten Ländern stellt eine große globale Herausforderung dar.
Stammzelltherapie
Die Transplantation von HLA-kompatiblen Stammzellen ist derzeit die einzige kurative Therapie für Hämoglobinopathien. Diese Methode erfordert jedoch einen passenden Spender, idealerweise ein HLA-identisches Geschwisterkind. Die Risiken umfassen Abstoßungsreaktionen und Infektionen, was die Therapie komplex und teuer macht.
In endemischen Ländern, wo die Prävalenz von Hämoglobinopathien hoch ist, stehen Patienten vor zusätzlichen Hindernissen. Der Zugang zu spezialisierten Transplantationszentren ist stark eingeschränkt, und die Behandlungskosten sind für viele Familien nicht zu bezahlen. Internationale Bemühungen sind noch notwendig, um diese Therapieoption in ressourcenarmen Regionen zur Verfügung zu stellen.
Um in das Thema noch tiefer einzusteigen und mehr Details zu den aktuellen Therapien und Fortschritten zu bekommen, bilde dich mit der online Fortbildung von Doctorflix "Hämatopoetische Stammzelltransplantation bei Hämoglobinopathien" weiter.
In endemischen Ländern, wo die Prävalenz von Hämoglobinopathien hoch ist, stehen Patienten vor zusätzlichen Hindernissen. Der Zugang zu spezialisierten Transplantationszentren ist stark eingeschränkt, und die Behandlungskosten sind für viele Familien nicht zu bezahlen. Internationale Bemühungen sind noch notwendig, um diese Therapieoption in ressourcenarmen Regionen zur Verfügung zu stellen.
Um in das Thema noch tiefer einzusteigen und mehr Details zu den aktuellen Therapien und Fortschritten zu bekommen, bilde dich mit der online Fortbildung von Doctorflix "Hämatopoetische Stammzelltransplantation bei Hämoglobinopathien" weiter.
7. Psychosoziale Aspekte
Häufig leiden Patienten unter sozialer Isolation, chronischen Schmerzen und Depressionen und die Familien sind durch die Erkrankung emotional und finanziell belastet.
Tipps für die Praxis:
Tipps für die Praxis:
- Verweise auf Selbsthilfegruppen und Beratungsstellen (z. B. Deutsche Gesellschaft für Hämatologie und Onkologie unter https://www.dgho.de). Zusätzlich können Patienten und ihre Familien von Plattformen wie "Orphanet" (https://www.orpha.net) profitieren, die umfassende Informationen zu seltenen Erkrankungen bereitstellen. Regionale Angebote wie die „Interessengemeinschaft Sichelzellkrankheit und Thalassämie“ (https://www.ist-ev.org/), vermitteln spezifische Anlaufstellen und Hilfen.
- Biete psychologische Unterstützung oder eine Überweisung an einen Psychologen an.
8. Zusammenarbeit mit Fachärzten
Eine enge Zusammenarbeit mit Hämatologen und spezialisierten Zentren ist essenziell. Als Haus- oder Kinderarzt kannst du diese Zusammenarbeit durch klare Überweisungswege und den Kontakt zu regionalen Netzwerken erleichtern. Beispielsweise kannst du Patienten an spezialisierte Ambulanzen für Hämoglobinopathien überweisen oder dich bei der Deutschen Gesellschaft für Hämatologie und Onkologie (DGHO) über geeignete Ansprechpartner informieren. Zudem ist der Austausch mit Transfusionsmedizinern und die Teilnahme an interdisziplinären Fallbesprechungen hilfreich, um eine umfassende Betreuung sicherzustellen.
Wann überweisen?
Wenn du bei Patienten unklare Symptome wie chronische Anämie, Schmerzen oder Entwicklungsstörungen bemerkst, solltest du genauer hinschauen. Insbesondere bei Verdacht auf eine Sichelzellkrise oder schwere Thalassämie ist es wichtig, den Patienten an einen Hämatologen oder in ein spezialisiertes Zentrum zu überweisen, damit eine gezielte Behandlung eingeleitet werden kann.
9. Fazit und praktische Tipps
Hämoglobinopathien sind komplexe Erkrankungen, die ein frühzeitiges Erkennen und eine gute Zusammenarbeit mit Spezialisten erfordern. Als Hausarzt kannst du durch gezielte Diagnostik und enge lebenslange Betreuung einen großen Beitrag zur Verbesserung der Lebensqualität leisten.
Checkliste für die Praxis:
- Diagnose: An Hämoglobinopathien denken bei Anämie, chronischen Schmerzen oder Migrationshintergrund.
- Prophylaxe: Impfungen und Antibiotikaprophylaxe.
- Therapie: Gezielte medikamentöse Behandlung und Transfusionen
- Betreuung: Regelmäßige Kontrolluntersuchungen und psychosoziale Unterstützung.
Auch wenn die Behandlung von Hämoglobinopathien komplex ist, kannst du durch dein Engagement und eine gute Zusammenarbeit mit Fachärzten entscheidend dazu beitragen, die Lebensqualität deiner Patienten zu verbessern. Die Zukunftsperspektiven mit neuen Therapien wie der Gentherapie sind vielversprechend, und es ist wichtig, weiterhin über Fortschritte informiert zu bleiben, z.B. durch unsere Fortbildung zu dem Thema "Herausforderungen bei der Versorgung von Erwachsenen mit Sichelzellkrankheit und Beta-Thalassämie".
Quellen
- Weatherall D. Hemoglobinopathies Worldwide: Present and Future. Current Molecular Medicine. 2008;8(7):592-599. doi:10.2174/156652408786241375
- Kohne E. Hemoglobinopathies. Deutsches Ärzteblatt International. Published online 8. August 2011. doi:10.3238/arztebl.2011.0532
- Naik RP, Smith-Whitley K, Hassell KL, et al. Clinical Outcomes Associated With Sickle Cell Trait: A Systematic Review. Ann Intern Med. 2018;169(9):619-627. doi:10.7326/M18-1161
- Gerber GF. Sichelzellanämie. MSD Manual Profi-Ausgabe. Published 9. April 2024. Zugegriffen Januar 30, 2025. https://www.msdmanuals.com/de/profi/h%C3%A4matologie-und-onkologie/h%C3%A4molytische-an%C3%A4mien/sichelzellan%C3%A4mie
- Gerber GF. Thalassämien. MSD Manual Profi-Ausgabe. Published 9. April 2024. Zugegriffen Januar 30, 2025. https://www.msdmanuals.com/de/profi/h%C3%A4matologie-und-onkologie/h%C3%A4molytische-an%C3%A4mien/thalass%C3%A4mien
- Gerber GF. Hämoglobin-S-Beta-Thalassämie. MSD Manual Profi-Ausgabe. Published 9. April 2024. Zugegriffen Januar 30, 2025. https://www.msdmanuals.com/de/profi/h%C3%A4matologie-und-onkologie/h%C3%A4molytische-an%C3%A4mien/h%C3%A4moglobin-s-beta-thalass%C3%A4mie
- AWMF-Leitlinie 025/016 „Sichelzellkrankheit“: 2. Auflage. Gesellschaft für Pädiatrische Onkologie und Hämatologie; 2020. Zugegriffen Januar 30, 2025. https://www.awmf.org/leitlinien/detail/ll/025-016.html
- Gerber GF. Sichelzellanämie. MSD Manual Profi-Ausgabe. Published 9. April 2024. Zugegriffen Januar 30, 2025. https://www.msdmanuals.com/de/profi/h%C3%A4matologie-und-onkologie/h%C3%A4molytische-an%C3%A4mien/sichelzellan%C3%A4mie#Pathophysiologie_v970288_de
- CasGevy | European Medicines Agency (EMA). European Medicines Agency (EMA). Zugegriffen Januar 30, 2025. https://www.ema.europa.eu/en/medicines/human/EPAR/casgevy
- Gieflelmann K. Sichelzellanämie und Beta-Thalassämie: erste CRISPR-Gentherapie zugelassen. Dtsch Arztebl International. 2023;120(49). Zugegriffen Januar 30, 2025. https://www.aerzteblatt.de/archiv/235618/Sichelzellanaemie-und-Beta-Thalassaemie-Erste-CRISPR-Gentherapie-zugelassen

Office Potsdam
Friedrich-Ebert-Straße 36
14469 Potsdam
Office Berlin
Schönhauser Allee 36
Haus 2 / Aufgang A
10435 Berlin
Hinweis: Die Inhalte auf Doctorflix sind ausschließlich für Ärzte und medizinisches Fachpersonal bestimmt. Sie dienen der fachlichen Fortbildung und sind nicht für Laien geeignet.
Gender-Hinweis: Zur besseren Lesbarkeit wird auf unserer Website das generische Maskulinum verwendet. Die in allen Texten verwendeten Personenbezeichnungen beziehen sich – sofern nicht anders kenntlich gemacht – auf alle Geschlechter.
Copyright © 2025 Doctorflix. Alle Rechte vorbehalten. Markenlogos nur zu Demonstrationszwecken.